| Kaz | Enfo | Ayiti | Litérati | KAPES | Kont | Fowòm | Lyannaj | Pwèm | Plan |
| Accueil | Actualité | Haïti | Bibliographie | CAPES | Contes | Forum | Liens | Poèmes | Sommaire |
Étude clinique
I. - INTRODUCTION
L'évolution générale de la drépanocytose est faite de périodes d'accalmie (état basal) entrecoupées de crises vaso-occlusives douloureuses pendant lesquelles une complication spécifique peut apparaître, qu'elle soit prévisible ou non.
Ces complications spécifiques apparaissent avec une électivité dans certaines tranches d'âge: c'est ce qui constitue l'évolution naturelle de la maladie. Les facteurs de décompensation douloureuse de la drépanocytose sont, classiquement et parmi. les plus fréquents:
- Les états de déshydratation même relatives (diarrhées, insuffisances d'apport hydrique).
- Toutes les situations où les besoins oxygénés se trouvent majorés (altitude, effort physique soutenu et inhabituel, voyages aériens sans pressurisation).
- L'acidose, notamment au cours des maladies infectieuses.
II. - LES COMPLICATIONS DE LA DREPANOCYTOSE
Le cours naturel de la maladie drépanocytaire laisse apparaître des complications dont certaines sont observées de façon préférentielle dans certaines tranches d'âge; l'apparition des premières complications cliniques aiguës s'observe vers l'âge de 6 ans, lorsque le taux d'HbF tend à rejoindre celui de l'adulte (Fig. 5).
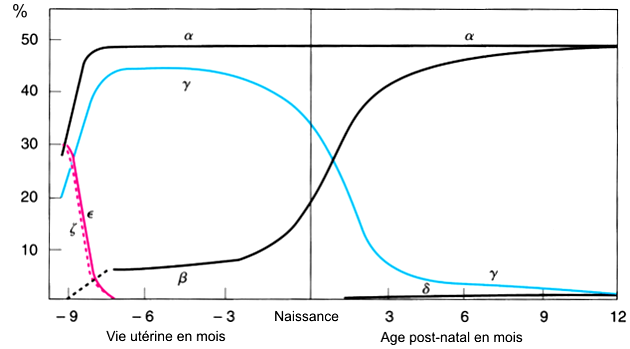
Fig. 5 : Évolution de la synthèse des chaînes de globine en fonction de l'âge.
La courbe de synthèse des chaînes α et γ-globine qui composent l'hémoglobine fœtale [2α, 2γ] coïncident avec un maximum entre 3 et 6 mois de vie utérine. La synthèse des chaînes γ amorce ensuite une chute qui devient quasi-complète 6 mois après la naissance: l'hémoglobine fœtale voit donc son taux diminué. À partir de la naissance, la synthèse des chaînes de β-globine subit une accrétion pour rejoindre les chaînes α demeurées élevées, et constituer l'hémoglobine A normale [9].
1. - À la naissance
Lorsque les antécédents familiaux ne sont pas précisés, rien ne permet de distinguer à la naissance un enfant drépanocytaire d'un autre. Les formes majeures de la maladie se prêtent toutefois assez aisément au diagnostic néonatal, par ponction sanguine au niveau du cordon ombilical [8].
2.- De 0 à 5 ans
La baisse progressive de l'hémoglobine foetale au profit de l'hémoglobine drépanocytaire ouvre la voie aux premières complications aiguës représentées par l'anémie, les crises vase-occlusives et l'infection.
2.1. - Les crises vaso-occlusives
Chez le nourrisson, les crises vaso-occlusives représentent les premières manifestations de la maladie. La présentation clinique la plus commune chez le jeune enfant est la dactylite aiguë ou syndrome pieds-mains. Elle se manifeste par un œdème inflammatoire d'un ou de plusieurs doigts, du dos des pieds et des orteils. Les traductions radiologiques de cette complication son retardées et donnent un aspect épaissi des corticales des os du carpe ou du tarse, un dédoublement périosté avec perte de substance. Ces signes régressent inconstamment avec une certaine tendance à la récidive.
La survenue d'une fièvre dans ce contexte suggère toujours une ostéite, ce qui motive le plus souvent une antibiothérapie parentérale entreprise dans l' attente des investigations complémentaires.
2.2. - L'anémie
Son début est précoce; elle apparaît vers l'âge de 4 mois principalement chez les homozygotes, avec un taux d'Hb entre 7 et 9 g/dl. C'est une anémie normochrome, normocytaire et régénérative. C'est le témoin d'une hyperhémolyse périphérique, d'autant qu'elle s'accompagne d'une hyperbilirubinémie libre avec chute de l'haptoglobine et augmentation du taux de réticulocytes.
À ce stade, la taille de la rate lorsqu'elle est précisée, devient par la suite un élément de surveillance clinique ultérieure. En effet, la rate du drépanocytaire involue vers l'âge de 7 ans (asplénie fonctionnelle), mais peut persister au-delà de cette limite chez les thalasso-drépanocytaires.
L'asplénie fonctionnelle est d'origine ischémique et peut être réversible, comme en témoignent des cas rapportés de reprise anatomo-fonctionnelle chez des homozygotes soumis à des protocoles transfusionnels au long cours [27].
2.3. - Les infections
Elles émaillent le cours évolutif de la drépanocytose; loin de ne représenter qu'un facteur de décompensation, elles rendent compte d'une certaine prédisposition et comportent un risque létal important. [63].
La fragilité aux infections est une caractéristique de la maladie drépanocytaire. Cette susceptibilité aux agents infectieux est la conséquence d'un infléchissement prouvé de la dynamique immunologique du drépanocytaire. C'est ainsi que des entérobactéries saprophytes tel edwarsiella tarda peuvent se trouver à l'origine d'infections sévères [75] chez le malade drépanocytaire. Plusieurs facteurs contribuent à cette susceptibilité aux infections:
- l'asplénie fonctionnelle qui prive progressivement l'organisme d'un composant immunologique fondamental dans l'enfance. Cette absence ouvre la voie à différents agents infectieux tels Streptococcus pneumonie, Neisseria meningitidis, Hæmophilus irifluenzæ et Salmonella [70].
- la baisse de l'activité phagocytaire impartie aux monocytes, ceux-là mêmes qui réagissent aux fractions C3b et Fe de la voie complémentaire alterne [70]. L'activité chimiotactique ainsi que la migration des polynucléaires neutrophiles sont elles aussi amoindries [14].
L'infection constitue dans le contexte de la maladie drépanocytaire un sujet de préoccupation constante, pouvant comporter des tableaux cliniques gravissimes avec des septicémies et atteintes neuro-méningées. On distingue les septicémies et les méningites purulentes, l'ostéomyélite et les complications respiratoires.
2.3.1. - Les septicémies et méningites pur lentes
Elles sont 30 fois plus élevées que dans la population générale; le pneumocoque est le germe le plus fréquemment en cause (70-80 % des cas). On rencontre aussi l'hémophilus et le méningocoque.
La population concernée est celle des enfants de moins de 4 ans (80 %). Entre 4 et 10 ans, le risque tombe à 10 %.
2.3.2. - Les ostéomyélites
L'incidence des ostéomyélites est élevée avant 2 ans, mais elles peuvent survenir à tous les âges. Elles concernent tous les os et réalisent des atteintes uni ou pluri focales. Les salmonelles sont les germes le plus souvent en cause [74], de même mais à une moindre fréquence les klebsielles [49, 54].
La clinique comporte une fièvre élevée qu'accompagnent une augmentation de la VS (> 20 mm), une hyperleucocytose (GB > 30’000 avec polynucléose neutrophile). Les hémocultures sont positives. La scintigraphie (technetium-gallium) montre une hyperfixation locale. Ces ostéomyélites peuvent s'abcéder (4e-5e jour) et laisser des séquelles osseuses.
2.3.3. - Les complications respiratoires
Ce sont les complications les plus fréquentes. Les germes en cause sont le pneumocoque [53], l'hémophilus et Mycoplasme pneumoniæ. Le tableau clinique associe une toux fébrile avec dyspnée et altération de l'état général. C'est le plus souvent une bronchopathie avec participation pleurale. Le diagnostic est affirmé les hémocultures, la ponction pleurale (si signes cliniques d'épanchement), la recherche des antigènes solubles et les examens sérologiques. La radiographie pulmonaire peut montrer un ou des foyers multiples. Le diagnostic différentiel est à faire avec l'infarctus pulmonaire.
Enfin, le traitement transfusionnel itératif, assez fréquent au cours de la drépanocytose, comporte le risque d'une contamination virale iatrogène (virus de l'immuno-déficience humaine, hépatites C et B, agent delta) [1, 22].
2.4. - La séguestration splénique aiguë
Décrite pour la première fois en 1945 par TOMLISON, cette situation d'urgence survient avec un maximum entre 6 mois et 2 ans [61]; inaugurée ou non par un facteur déclenchant reconnu, elle se caractérise par une aggravation brutale de l'anémie (le taux d'Hb chute entre 5 et 3 g/dl) et par une augmentation soudaine du volume de la rate au-dessus de ses mensurations de référence.
Le volume abdominal est donc augmenté. L'anémie aiguë, brutale, avec une chute importante du taux d'hémoglobine entraîne une anoxie cérébrale, ce qui peut générer des troubles comportementaux. La séquestration splénique s'explique par une rétention sanguine au niveau des sinusoïdes spléniques pouvant conduire au collapsus cardio-vasculaire. En clinique, la survenue d'une crise aiguë de déglobulisation fait discuter:
- la séquestration splénique,
- la crise hémolytique aiguë infectieuse (virus d'Epstein-Barr, mycoplasme, cytomégalovirus),
- l'érythroblastopénie aiguë due au parvovirus B19.
Le risque de séquestration splénique touche les homozygotes jusque vers l'âge de 6 ans. Au-delà, la rate involue mais reste en revanche fonctionnelle chez les doubles hétérozygotes S/b-Thal, SIC et S/PHFIF chez qui les récidives restent une préoccupation.
Le traitement de la crise de séquestration splénique est transfusionnel. La splénectomie est envisagée en cas de récidive car le risque mortel croît avec les épisodes ultérieurs (de 12 % au cours de la première crise à 20 % pendant les suivantes). Ces récidives sont particulièrement à craindre dans les associations possibles de la drépanocytose avec une microsphérocytose héréditaire [72].
3. - De 5 ans à l'adolescence
On peut à ce stade dépister les premiers signes d'altération organique; c'est aussi la période des complications vasculaires neurologiques.
3.1. - Complications neuro-vasculaires
Les accidents vasculaires cérébraux (AVC) représentent la complication aiguë majeure au cours de l'évolution de la drépanocytose. Le premier cas d'AVC fut rapporté en 1923 par SYDENSTRICKED [67] à propos d'un garçonnet de 6 ans, chez lequel un antécédent d'hémiparésie gauche à l'âge de 4 ans avait été consigné.
Deux autres cas furent signalés entre 1923 et 1935, chez deux jeunes enfants de 7 et 6 ans, avec pour le premier un état de coma complétant un tableau d'hémiparésie gauche, suivi de décès, avec hémorragie sous-arachnoïdienne et ramollissement droit confirmés par l'autopsie, et pour le second un déficit hémiparétique gauche laissant des séquelles motrices [4]. Ces enfants étaient drépanocytaires homozygotes avec des passés de crises douloureuses à répétition.
L'anémie, quelle qu'en soit l'étiologie, entraîne une élévation du débit cardiaque. Au niveau des vaisseaux cérébraux, cela se traduit par une dilatation du lit vasculaire secondairement à l'hyper-perfusion cérébrale, laquelle est inversement proportionnelle à l'hématocrite [57]. Nous avons donc ici un premier facteur d'agression mécanique, barométrique.
D'autre part, chez les drépanocytaires, la répétition des microthrombi au niveau des vaisseaux de faible calibre (dont les vasa-vasorum) entretient une agression anoxique qui provoque des remaniements tissulaires, avec comme conséquence l'apparition d'une néovascularisation et d'une hyperplasie endothéliale, autant d'éléments de fragilisation pariétale. Ces remaniements réduisent le calibre des vaisseaux cérébraux ou favorisent la formation de poches anévrismales.
La fragilisation vasculaire avec formation d'anévrismes explique la survenue d'accidents cérébraux de type hémorragique; la réduction du calibre des petits vaisseaux pal- l'hyperplasie endothéliale est à l'origine d'incidents ischémiques.
Les AVC surviennent préférentiellement chez les sujets homozygotes S/S et à un moindre degré chez les doubles hétérozygotes S/C. Les doubles hétérozygotes S/β-thal en sont pratiquement exempts. L'âge moyen d'exposition est de 7,7 ans. 5 à 17 % des drépanocytaires développeront un AVC, avec une prédominance des accidents ischémiques (75 %) sur les accidents hémorragiques (25 %) [56, 69].
Les accidents hémorragiques sont de survenue plus tardive vers l'âge de 25 ans. Les facteurs de prédiction de cette complication sont inconnus. Les AVC au cours de la drépanocytose correspondent à diverses lésions cérébro-vasculaires:
- infarctus cérébraux: observés surtout dans les génotypes homozygotes SIS et dans l'enfance; les récidives se voient dans 67 % des cas au cours des trois années qui suivent l'épisode initial et majorent les séquelles préexistantes (motrices, intellectuelles).
- hémorragies sous-arachnoïdiennes et intracérébrales: elles sont plus tardives (entre 14 et 36 ans); les tableaux cliniques sont. plus spectaculaires, entraînant coma et décès dans certains cas. La population la plus exposée est celle des homozygotes S/S.
- embolies graisseuses disséminées: un cas rapporté dans notre littérature chez un adolescent de 14 ans, double hétérozygote S/C décédé au décours d'un tableau d'infarctus osseux, dont l'autopsie avait révélé des embolies graisseuses pulmonaires, rénales et cérébrales [56].
Les accidents neuro-vasculaires répondent bien au traitement transfusionnel en phase aiguë sous forme d'échanges transfusionnels partiels (ou exsanguino-transfusion partielle). Mais ce type de complication qui menace le pronostic vital, ouvre surtout la discussion sur la possibilité d'une greffe médullaire génoidentique.
3.2. - Altérations hépato-vésiculaires
Les lithiases biliaires sont habituelles au cours de la drépanocytose; dans la moitié des cas, ce sont des lithiases pigmentaires dues à la chronicité de l'hémolyse. Leur occurrence n'est pas obligatoire. Elles peuvent survenir très tôt chez l'enfant ou plus tard après l'âge de 20 ans. Les calculs peuvent être de nature cholestérolique, traduisant des anomalies du métabolisme de l'acide biliaire couplées à un dysfonctionnement hépato-vésiculaire [25].
Les calculs biliaires concernent les génotypes SIS (22,6 %) et SIC (33,3 %) selon l'étude qui nous sert de référence [19]; les lithiases se rencontrent volontiers chez le drépanocytaire adulte (entre 22 et 37 ans).
Leur existence chez des sujets plus jeunes devient un argument en faveur d'autres mécanismes étio-pathogéniques que la seule chronicité de l'hémolyse qui génère des lithiases pigmentaires.
Des études sur des pièces autopsiques ou opératoires chez les jeunes enfants lithiasiques pourraient faire la lumière sur la nature biochimique des lithiases retrouvées, et préciser un mécanisme différent dans cette tranche d'âge.
Les lithiases vésiculaires occasionnent des complications (cholécystites infectieuses) difficiles à différencier des crises abdominales vaso-occlusives. Pour cette raison, certains auteurs préconisent une surveillance échographique régulière, et le cas échéant une cholécystectomie réglée, à froid après échange transfusionnel pour éviter la survenue d'infections vésiculaires et diminuer les crises douloureuses abdominales [17].
3.3. - Manifestations oculo-conjonctivales
Ce sont des remaniements fragilisants qui associent sinuosité vasculaire et dilatations micro-anévrismales; ces lésions sont visibles au biomicroscope. Elles sont dues à l'infiltration pariétale par des drépanocytes. Leur importance tient à l'âge du malade et à la sévérité de la maladie, mais elles ne semblent pas entraîner de complication notable, si ce n'est une fragilisation conjonctivale. Ce n'est qu'un aspect particulier des modifications micro-vasculaires générales dues à la drépanocytose.
3.4. - Atteintes iriennes
Il n'existe pas d'uvéite typique de la maladie drépanocytaire; la fragilité des malades drépanocytaires devant l'infection peut les exposer à une uvéite commune. Cependant les drépanocytaires ont la particularité d'entretenir des uvéites chroniques évoluant à bas bruit et relevables par des manœuvres de massage oculaire qui font apparaître le signe de Tyndall.
3.5. - Rétinopathies
Elles sont de type prolifératif et augmentent de fréquence avec l'âge du malade. La rétinopathie drépanocytaire concerne les homozygotes S/S (40 %), mais surtout les doubles hétérozygotes S/C (70 % ) [18] avec, dans cette population, une certaine électivité masculine.
Les doubles hétérozygotes S/b-Thal ne sont pas épargnés, malgré une faible documentation à ce sujet. Les facteurs prédictifs du risque sont d'ordre hématologique et ne comportent pas de prédisposition familiale [29].
Les facteurs de prédiction de la rétinopathie drépanocytaire sont: e un taux d'Hb élevé (sujets mâles homozygotes S/S).
- un taux bas d'HbF: sujets homozygotes SIS des deux sexes, sujets féminins hétérozygotes SIC.
- un VGM élevé, toutes causes confondues (carence en folates, taux élevé d'érythroblastes): sujets S/C des deux sexes.
Les rétinopathies drépanocytaires sont souvent bilatérales, symétriques et suivent une classification évolutive (examen au verre à trois miroirs, angiographie à la fluorescéine) (Tab. 5). Une classification simplifiée, celle de HAMARD, résume les altérations rétiniennes en deux stades:
- rétinopathie non compliquée où les lésions rétiniennes sont périphériques et équatoriales.
rétinopathie compliquée avec atteinte du pôle postérieur (papille, macula, vitrée) ou décollement rétinien.
Tab. 5. - Rétinopathie drépanocytaire (d'après Goldberg).
| Degrés d'atteintes rétiniennes | Aspects de la rétine. |
| stade 1 | ischémie hémorragies avec séquelles pigmentées. |
| stade 2 | apparition d'anastomoses périphériques |
| stade 3 | proliférations capillaires |
| stade 4 | hémorragies au niveau de la néovascularisation capillaire. |
| stade 5 | décollement rétinien, cécité. |
3.6. - Troubles cardio-vasculaires
Il est habituel de trouver chez le drépanocytaire un souffle précordial éjectionnel mimant une insuffisance valvulaire. Ces manifestations sont fonctionnelles et relèvent de l'anémie.
En dépit de cela, il n'existe à proprement parler pas de cardiopathie spécifiquement drépanocytaire qui correspondrait à une entité pathogénique distinguable du cœur anémique [10]. En définitive, la drépanocytose est un cause de cardiopathie anémique. Cependant au cours de la drépanocytose, on observe bien des modifications échocardiographiques tant fonctionnelles qu'anatomiques, avec dans 2/3 des cas une hypertrophie ventriculaire gauche excentrique, un index cardiaque augmenté et une diminution des résistances périphériques. Dans - le 1/3 restant, il existe une altération de la fonction pompe du ventricule gauche.
Ces anomalies s'accentuent avec l'âge, mais ne débouchent pas, en l'absence de facteurs étrangers à la drépanocytose, sur l'insuffisance cardiaque congestive [24].
3.7. - Manifestations osseuses
Les atteintes osseuses sont absentes chez le nourrisson de moins de 6 mois en raison d'une persistance de l'HbF; les altérations osseuses sont plus fréquemment observées parce que l'espérance de vie des drépanocytaires s'est considérablement améliorée et ceci grâce à une prise en charge efficace.
Les traductions radiologiques de l'ostéopathie drépanocytaire ont parfois des caractéristiques spécifiques. Les remaniements osseux sont dus à l'hyperplasie médullaire d'entraînement par l'anémie, l'ischémie et une greffe infectieuse toujours possible; sur le plan radiologique, on observe les aspects suivants:
- agrandissement des cavités,
- raréfaction de la trame osseuse,
- amincissement cortical.
Les deux derniers aspects simulent donc véritablement une ostéoporose. Ces aspects sont moins fréquemment observés chez les drépanocytaires vivant en Europe et aux Etats-Unis que chez ceux d'Afrique.
Sur le plan clinique, les modifications osseuses s'observent au cours des crises d'ischémie et d'infarctus osseux plus précoces chez les homozygotes. La clinique associe presque toujours douleur et gonflement des parties molles. L'apparition d'une fièvre avec hyperleucocytose pose toujours le problème du diagnostic différentiel avec une ostéomyélite.
Les crises douloureuses osseuses correspondent à l'ischémie ou à un infarctus osseux; le syndrome mains-pieds de l'enfant en est une traduction particulière.
L'infarctus osseux intéresse surtout la diaphyse des os longs et reproduit des signes radiologiques retardés (une à deux semaines), sous forme d'ossification périostée localisée à la jonction métaphyso-épiphysaire.
Les métaphyses présentent en dualité des zones condensées et des zones de radio-transparence. Le diagnostic différentiel est mal aisé à faire avec une ostéomyélite.
L'infarcissement épiphysaire semble plus fréquente chez les doubles hétérozygotes S/C adultes; les lésions sont bilatérales et intéressent volontiers les têtes fémorales et humérales aboutissant à des nécroses céphaliques souvent asymptomatiques, ce qui explique la fortuité de leur découverte.
À côté de la radiographie conventionnelle, la scintigraphie isotopique au technétium 99m phosphaté confirme l'infarcissement osseux (hypofixation) ou l'hyperplasie médullaire (hyperfixation).
Les troubles osseux, associés aux carences alimentaires observées dans les zones d'endémie drépanocytaire, ont à long terme un retentissement péjoratif sur la croissance staturo-pondérale [20].
3.8. - Atteintes rénales
Les atteintes rénales sont la conséquence des modifications hémodynamiques liées à l'anémie, et des phénomènes vaso-occlusifs à l'intérieur de la médullaire rénale.
- L'hyposthénurie: c'est le pouvoir insuffisant de concentration des urines. D'apparition précoce, elle devient irréversible à l'âge adulte.
- Les hématuries: elles sont le plus souvent totales et unilatérales. Elles sont de résolution spontanée, avec une tendance à la récidive. Les hématuries sont le plus souvent dues à une nécrose papillaire.
- Le syndrome néphrotique: il fait suite à une protéinurie devenue permanente. Il peut s'agir d'un syndrome néphrotique typique (anasarque, hypercholestérolémie, hypoalbuminémie et protéinurie abondante) [30] dont l'aggravation conduit à l'insuffisance rénale.
- Les infarctus rénaux: la médullaire rénale y est exposée, du fait des conditions locales prédisposant à la falciformation (taux bas d'O2, acidité, hyperosmolarité, stase circulatoire et ischémie chronique.
III. - LE CAS DES DRÉPANOCYTAIRES HÉTÉROZYGOTES
La drépanocytose hétérozygote ou trait drépanocytaire (A/S) est en règle générale asymptomatique. Les caractéristiques hématimétriques du sang périphérique des drépanocytaires hétérozygotes sont identiques à celles du sang normal [35]. Cependant, quand les globules rouges sont incubés en milieu désoxygéné (test d'Emmel), le phénomène de falciformation se manifeste et fait apparaître des drépanocytes. Il est donc certain que l'anoxie favorise la falciformation des hématies A/S.
On décrit chez les hétérozygotes, essentiellement des hématuries facilement réduites par l'hydratation, éventuellement l'acide ε-amino-caproïque et rarement des nécroses papillaires [6]. Enfin, survenant à haute altitude, infarctus spléniques et pulmonaires.
En conclusion, le drépanocytaire hétérozygote devient symptomatique lorsque les conditions habituelles de falciformation sont rassemblées: hypoxie (au cours des infections pulmonaires sévères, l'altitude), acidose et déshydratation gave.
IV. - CONCLUSION
Au terme de cette étude, la drépanocytose apparaît comme une maladie hématologique chronique à retentissement organique large. La précocité et la régularité de la prise en charge médicale permettent de minimiser les altérations organiques induites par cette maladie et donc d'en améliorer le pronostic évolutif. Dans ces conditions, l'allongement de l'espérance de vie laisse malheureusement apparaître des complications dégénératives invalidantes.
Diagostic positif de la drépanocytose
La maladie est suspectée devant une anémie normochrome, normocytaire, régénérative (numération et formule sanguines); le diagnostic positif de la drépanocytose repose sur la mise en évidence de l'hémoglobine S à l' électrophorèse de l'hémoglobine. Une série de tests de laboratoire utilisant les propriétés de l'HbS permettent ensuite de confirmer ce diagnostic.
1. - Numération et formule sanguines
- Taux d'Hb compris entre 7 g/dl et 9 g/dl,
- volume globulaire moyen entre 95 et 95 µm3,
- teneur globulaire moyenne en Hb normale,
- augmentation du taux des réticulocytes (5 à 3 %).
2. - Le frottis sanguin
- Anisocytose,
- présence de cellules cibles,
- polychromatophilie,
- parfois présence de drépanocytes irréversibles.
3. - Le test de falciformation (test d'Emmel)
C'est l'examen microscopique de cellules spontanément désoxygénées entre lame et lamelle.
4. - Les tests de solubilité
Ces tests dérivent de la méthode proposée par ITANO en 1953, fondée sur la solubilité très diminuée en tampon phosphate concentré d'une solution d'HbS désoxygénée par le dithionite. L'introduction d'agents hémolysants dans le tampon permet de pratiquer ce test directement sur du sang. Après centrifugation de l'hémolysat, on obtient un culot et un surnageant dont l'examen permet ou non de confirmer la présence d'hémoglobine S et de définir une présentation hétéro ou homozygote:
- surnageant rouge + dépôt blanc de protéines: solubilité normale, sujet indemne.
- surnageant rose + dépôt d'hématies et de protéines: solubilité diminuée, S hétérozygote.
- surnageant pâle + dépôt de toutes les hématies avec des protéines: insolubilité totale, S homozygote.
5. - L'électrophorèse de l'hémoglobine
5.1. - Méthode
L'électrophorèse se pratique sur support d'acétate de cellulose en pH basique, ce qui donne des résultats rapides en quelques minutes. L'hémoglobine S migre entre les hémoglobines A et A2. Le test électrophorétique employé seul demeure insuffisant, car il ne permet pas toujours de déceler tous les mutants qui présentent une différence de charge électrique identique à l'HbS. Ce sont, par exemple, les hémoglobines D Punjab et Lepore qui peuvent donner des associations cliniquement comparables à la drépanocytose homozygotisme S/S. Pour caractériser ces mutants, on pratique le test de focalisation isoélectrique. L'électrophorèse en milieu acide peut aussi aider à différencier ces différents mutants.
5.2. - Les résultats électrophorétiques
Dans la drépanocytose homozygote, l'hémoglobine S est majoritaire, l'HbA2 est sensiblement normale et l'HbF est présente à des taux variables. À côté de la forme homozygote, on peut rencontrer:
- une S/β-thalassémie: étude familiale, microcytose, augmentation franche de l'HbA2, l'HbF est présente à un taux variable.
- une S/β+-thalassémie,
- un trait drépanocytaire A/S où le taux d'HbS environ 40 %,
- l'association à une α-thalassémie: microcytose familiale, faible expressivité de l'HbS,
- l'association S/C: l'hémoglobine drépanocytaire représente 50 % du total.
6. - Évaluation biologique complémentaire
Une fois le diagnostic positif établi, la quantification de l'hémoglobine S est utile, compte tenu de l'importance physiopathologique de la concentration de l'HbS dans le globule rouge. Les constantes hématimétriques constituent une donnée importante.
Au cours de la prise en charge d'un drépanocytaire, de nombreux facteurs d'interaction sont à surveiller, tels l'acide folique et le fer, directement liés à l'anémie. Nous le verrons, la carence en acide folique est le précurseur d'une érythroblastopénie aiguë. La surveillance du fer sérique permet de dépister des hyposidérémies qui pourraient aggraver l'anémie drépanocytaire. Le traitement transfusionnel n'expose pas ici au risque de surcharge ferrique, car la méthode utilisée est celle des échanges transfusionnels.
La transfusion conventionnelle est encore utilisée dans les pays en voie de développement, mais en nombre limité compte tenu des moyens exigés par ce type de traitement. Elle est également utilisée lors des crises aiguës de déglobulisation. Dans tous les cas, il faut prendre en compte et surveiller les conséquences possibles de la thérapeutique transfusionnelle (allo-immunisation, contamination infectieuse).
Certains éléments dosables revêtent un intérêt pronostique; c'est le cas de l'hémoglobine F dont le rôle modérateur est bien connu en pathologie drépanocytaire.
Diagnostic ante et néonatal
I. - GÉNÉRALITES
Le pronostic évolutif du syndrome drépanocytaire est très favorablement influencé par la précocité du dépistage et de la prise en charge des personnes malades ou à risque. C'est ici que les diagnostics ante et néonatal, de même que le conseil génétique retrouvent toute leur importance.
Le conseil génétique dont le rôle est principalement informatif et éducatif, s'aide du dépistage des sujets à risque, ainsi que des diagnostics ante et néonatal. Le dépistage drépanocytaire ne s'identifie pas à une prévention de type primaire dont le but serait de réduire l'incidence des syndromes drépanocytaires: il informe mais laisse le choix aux parents de décider de leur descendance.
II. - DIAGNOSTICS ANTE ET NÉONATAL
1. - Le diagnostic anténatal
Il peut correspondre à une démarche médicale prénuptiale lorsque les futurs parents sont sensibilisés et se savent porteurs sains ou appartenant tous les deux à des populations à risque. Mais dans la plupart des cas, le diagnostic anténatal intervient alors qu'une gestation est déjà engagée, dans une famille comprenant un ou des membres drépanocytaires.
Dans le contexte d'une première grossesse, les parents ont rarement un suivi médical spécifique antérieur et donc «ne se connaissent pas». Ils ont recours au diagnostic anténatal pour les naissances suivantes.
La recherche anténatale de la drépanocytose n'est d'aucun apport lorsque les parents sont homozygotes: la descendance est dans ce cas obligatoirement homozygote et donc malade. Hormis ce cas particulier, le diagnostic anténatal est indiqué dans les situations suivantes:
- risque d'une descendance homozygote (parents doubles hétérozygotes ou porteurs du trait drépanocytaire),
- risques d'une association composite S/β-thalassémique, S/D Punjab ou S/O Arab.
1.1. - La conduite du diagnostic anténatal
L'étude de l'hémoglobine se fait à partir d'un prélèvement sanguin ou de cellules nucléées fœtales. Le prélèvement de sang fœtal se pratique au second trimestre (18e semaine). À ce stade la cavité amniotique a une taille suffisante et les vaisseaux un calibre suffisant pour permettre la ponction, sous contrôle échographique. À cette période, la synthèse des chaînes de 13-globine est suffisante.
À partir des cellules nucléées fœtales prélevées, on étudie l'ADN fœtal, entre la 16e et la 18e semaine. Le produit recueilli par amniocentèse donne après centrifugation un culot de cellules fibroblastiques dont on explore l'ADN à la recherche d'une mutation.
Les deux méthodes présentent l'inconvénient de fournir des résultats tardifs, peu compatibles avec l'éventualité d'une option pour l'intervention de grossesse.
La recherche anténatale de la drépanocytose n'est d'aucun apport lorsque les parents sont homozygotes: la descendance est dans ce cas obligatoirement homozygote et donc malade. Hormis ce cas particulier, le diagnostic anténatal est indiqué dans les situations suivantes:
- risque d'une descendance homozygote (parents doubles hétérozygotes ou porteurs du trait drépanocytaire),
- risques d'une association composite S/β-thalassémique, S/D Punjab ou S/0 Arab.
Tab. 6. - Techniques de prélèvement pour diagnostic prénatal et risque de fausse couche observé [37].
| Technique | Terme* | Risque fœtal (%) |
| Biopsie de trophoblaste (choriocentèse) | à partir de 10 semaines | 2 à. 4 |
| Ponction de liquide amniotique (amniocentèse) | à partir de 17 semaines | 0,5 |
| Ponction de sang fœtal (fætoscopie) | à partir de 18 semaines | 2 |
* Le terme est exprimé en semaines d'aménorrhée à partir du premier jour des dernières règles avec confirmation échographique.
C'est pour cette raison qu'on peut aussi entreprendre la biopsie de cellules trophoblastiques à partir des villosités choriales dont la taille permet dès la 10ème semaine un bon repérage échographique.
2. - Le diagnostic néonatal
II permet de reconnaître précocement le profil sanitaire d'enfants naissant de familles à risque. Il s'intègre donc au cadre général du dépistage des anomalies génétiques transmissibles, lorsque la notion d'un risque est établie (données épidémiologiques, antécédents familiaux).
Le diagnostic néonatal se pratique à partir de prélèvements sanguins réalisés au niveau du cordon ombilical, ou par ponction capillaire en microtube (vers le 3e jour). Il permet, une fois le diagnostic confirmé, d'organiser le suivi des malades, avec la mise en place d'un programme vaccinal, la prophylaxie antipneumococcique, l'éducation sanitaire et le conseil génétique à dispenser aux parents [32].
III. - TRANSMISSION DU GÈNE DRÉPANOCYTAIRE
Le gène drépanocytaire se transmet selon le mode autosomique codominant. En pratique, cela n'a pas beaucoup de signification pour des parents inquiets d'être informés sur leur descendance. On peut donc leur fournir des informations simples, en terme de probabilité (exprimée en pourcentage) d'avoir ou non des enfants malades. Cela se réalise à l'aide d'un classique tableau de croisement de Mendel.
Nous avons envisagé dans le tableau n° 7 les cas de croisements les plus courants avec comme critère de base la présence du gène drépanocytaire chez au moins un des deux parents. Nous avons défini un «profil» pour chaque type de croisement, nommés de A à J.
Il en ressort un tableau synthétique qui donne des indications sur la génétique des enfants à partir des parents, sous la réserve que le génotype de ces derniers soit connu. La notation [S/βxA/S], par exemple, signifie le croisement d'un thalasso-drépanocytaire avec un sujet porteur du trait drépanocytaire.
Tab. 7 - Evaluation du risque drépanocytaire.
Profil |
Signification (descendance) |
Croisements correspondants |
Descendance indemne du gène b-2 % |
Descendance non drépanocytaire1 |
A |
100% trait drépanocytaire |
[A/AxS/S] |
0 |
100 |
B |
50% trait drépanocytaire |
[A/AxA/S] |
50 |
100 |
C |
25% trait drépanocytaire |
[A/SxS/β] |
25 |
50 |
D |
25% homozygote |
[A/SxA/S] |
25 |
75 |
E |
50% homozygote |
[S/SxS/β] |
0 |
0 |
F |
100% homozygote |
[S/SxS/S] |
0 |
0 |
G |
25% dh2 S/b-thal |
[A/SxS/β] |
0 |
0 |
H |
50% dh2 S/b-thal |
[S/βxS/β] |
0 |
0 |
I |
25% dh2 S/C |
[A/SxS/C] |
25 |
50 |
J |
50% dh2 S/C |
[S/SxS/β] |
0 |
0 |
Ces résultats sont utiles lorsque le génotype des parents peut âtre déterminé. Ils ne concernent que la maladie drépanocytaire. Toutes les possibilités de croisement étant envisagées, le pourcentage des descendants non drépanocytaires inclut des combinaisons non documentées dans leurs présentations cliniques (25% de descendants β/C pour le croisement [S/SxS/C]), ou malades du fait d'une thalassémie majeure (25% d'enfants β/β dans les croisements [S/βxS/β].
IV. - CONCLUSION
Le pronostic de la drépanocytose dépend de la précocité du diagnostic ainsi que de la qualité et de la régularité du suivi ultérieur. Les drépanocytaires arrivant à l'âge adulte sont ceux qui ont pu bénéficier de ces conditions. En Afrique (continent d'endémie drépanocytaire), la prise en charge des malades reste le plus souvent limitée au traitement des crises douloureuses en hôpital général, sans suivi ultérieur entre deux épisodes critiques.
Les raisons de cette insuffisance médicale sont d'ordre économique et dépendent du niveau sanitaire (en terme d'équipement) général des pays. En l'absence d'information suffisante, les patients drépanocytaires des zones d'endémie demeurent pour la plupart dans la méconnaissance de tout ce qui sur le plan clinique peut correspondre à un facteur de décompensation, aux prémices d'une transformation critique susceptible de déboucher sur des complications toujours imprévisibles tant dans leur nature que leur gravité.
Les drépanocytaires qui se rapprochent d'un pays médicalement développé (Europe, États-Unis) connaissent pour certains une meilleure évolution de leur maladie. Cependant, le seul fait de parvenir dans un pays médicalement développé n'est pas suffisant en soi, car les malades arrivent avec leurs particularités culturelles et leurs croyances.
Le niveau d'alphabétisation des personnes est variable et dans certains cas, à cause de la barrière linguistique, le conseil génétique et l'éducation sanitaire réservés au drépanocytaire et à son entourage ne peuvent être efficacement prodigués.
Enfin, il faut admettre qu'une prise en charge durable et suivie demande que les malades soient bénéficiaires d'une couverture sociale, et qu'il puissent accéder à des services médicaux spécialisés dédiés à leur maladie.
La drépanocytose constitue une lourde charge à gérer pour les familles, qui ont besoin en plus de l'assistance médicale, d'un soutient psychologique permanent. Ce soutient intervient plus particulièrement, lorsqu'après le diagnostic anténatal, une interruption de grossesse a été réalisée. La femme africaine, par exemple, conçoit la maternité comme un accomplissement nécessaire de sa féminité. De cette façon, la perspective d'un recours ultérieur à l'avortement pour les mêmes raisons peut la conduire à éluder les indications du conseil génétique et à se soustraire à toute surveillance dans l'espoir «désespéré» d'une grossesse normale. Un autre aspect de la question concerne les couples d'homozygotes informés de leur descendance. Le problème qui se pose alors est celui du choix de demeurer sans enfants ou avec des enfants tous malades. Cette situation est généralement une menace pour la survie du couple et justifie un suivi psychologique des familles.
SOMMAIRE | HISTORIQUE | RAPPELS | ÉPIDÉMIOLOGIE |ÉTUDE CLINIQUE | DIAGNOSTIC POSITIF DE LA DRÉPANOCYTOSE | DIAGNOSTIC ANTE ET NÉONATAL | TRAITEMENT DE LA DRÉPANOCYTOSE | LES PERSPECTIVES THÉRAPEUTIQUES DE LA DRÉPANOCYTOSE | CONCLUSION | BIBLIOGRAPHIE