| Kaz | Enfo | Ayiti | Litérati | KAPES | Kont | Fowòm | Lyannaj | Pwèm | Plan |
| Accueil | Actualité | Haïti | Bibliographie | CAPES | Contes | Forum | Liens | Poèmes | Sommaire |
Introduction
Nous gardons le souvenir d'enfants dont nous avons partagé la tranche d'âge, en République Centrafricaine. Ceux-ci se reconnaissaient à leur maigreur, à leur teint crayeux et pour les plus grands, à une desquamation de la peau au niveau des jambes. C'est à cela que se distinguaient ceux qu'on avait pris l'habitude de nommer avec les deux premières syllabes du mot «drépanocytaire».
L'existence de ces enfants était partagée entre la certitude d'une prochaine hospitalisation et des tentatives désespérées de scolarisation. Ils avaient la réputation d'une longévité compromise et une vie relationnelle pauvre; c'est ainsi que progressivement, on les perdait de vue. L'intérêt que nous avons porté à ce travail résulte en partie de ce vécu.
La drépanocytose est une pathologie répandue en Afrique où elle pose d'importants problèmes sanitaires. Son intrication avec les pathologies infectieuse et parasitaire lui confère une gravité particulière, en contribuant à un taux de mortalité infantile élevé.
En zone d'endémie drépanocytaire il n'existe pratiquement pas, pour des raisons économiques, de système de prise en charge diagnostique et thérapeutique orienté vers le long terme. C'est ainsi que la plupart des enfants atteints de cette maladie n'arrivent pas à l'âge adulte. Il nous a donc paru intéressant de relever par l'étude de dossiers médicaux, les particularités cliniques et thérapeutiques actuelles de la maladie, ainsi que le bénéfice à. escompter d'une prise en charge suivie, lorsqu'elle est accessible.
En nous aidant des données actualisées de la littérature, nous avons souhaité approfondir le sujet de la maladie drépanocytaire dans des domaines aussi variés que ceux de la génétique, de l'épidémiologie et des perspectives thérapeutiques. Enfin, à travers ce travail, nous aurons cherché à désigner une pathologie mondialement rencontrée, riche en enseignement lourde de conséquences.
Prépondérante par sa fréquence, la drépanocytose est l’hémoglobinopathie qui n'a néanmoins suscité d'intérêt qu'au début du siècle par des cas observés chez les Noirs américains.
Chapitre I : HISTORIQUE
- 1910: HERRICK remarque chez un étudiant jamaïcain de Chicago la présence de globules rouges déformés en faucille; la maladie tient son nom de cette particularité morphologique: anémie falciforme, drépanocytose (du grec, drepanos = faucille).
- 1917: EMMEL évoque le caractère familial de la tare.
- 1927: HAHN et GILLESPIE constatent que la déformation globulaire ou falciformation est une conséquence de la désaturation en oxygène, précisément lorsque la pression partielle artérielle en oxygène (PaO2) baisse au-dessous de 50 millimètres de mercure. La falciformation est un phénomène réversible.
- 1933: DIGGS répartit les drépanocytaires en deux catégories cliniques distinctes, d'une part les patients asymptomatiques et d'autre part ceux présentant spontanément un état anémique aggravé.
- 1947: NEEL distingue à partir d'études familiales deux formes génétiques: hétérozygote correspondant au trait drépanocytaire et homozygote correspondant à la forme symptomatique. En effet les sujets anémiques avaient le plus souvent des parents sains. Ces deux formes génétiques (parents sains, descendance malade) s'accordent avec la classification de DIGGS.
- 1949: PAULING, ITANO, SINGER et WELLS constatent une différence de migration électrophorétique entre l'hémoglobine normale adulte et l'hémoglobine drépanocytaire, découvrant ainsi la drépanocytose comme une maladie primitivement moléculaire. L'hémoglobine drépanocytaire est nommée S (HbS).
- 1950: HARRIS décrit la formation d'un gel par désoxygénation d'une solution concentrée d'HbS; dans la même période, PERUTZ et MITCHISON objectivent la moindre solubilité de la désoxy-HbS dans des conditions comparables de désaturation en oxygène. Cette propriété est à l'origine des manifestations cliniques de la drépanocytose.
- 1956-1959: INGRAM analyse la lésion moléculaire au niveau de l'hémoglobine; de cette étude viendra la compréhension du comportement électrophorétique de la molécule d'HbS. Comparé à son homologue normal, l'hémoglobine S comporte une anomalie dans l'ordre d'enchaînement des acides aminés constitutifs: à la place attendue d'un acide glutamique (position 6 sur la chaîne bêta) est retrouvé un résidu valine (β6: Glu → Val).
Il sera ensuite démontré (en 1960) que cette substitution résulte de la mutation au niveau du triplet ADN codant guanine-adénine-guanine (G-A-G) en guanine-thymine-guanine (G-T-G).
La drépanocytose entretient de nos jours un intérêt sans cesse accru, à cause de sa prévalence, son extension et les conséquences qu' elle implique en santé publique. De nombreux travaux de recherche sont consacrés à cette maladie, qui permettent une amélioration notable de l'espérance de vie et du confort des malades.
![]()
![]()
![]()
Chapitre II : RAPPELS
I. - L'HÉMOGLOBINE ADULTE NORMALE
1. - Structure :
L'hémoglobine est une hétéroprotéine qui associe l'hème (constituant non protéique) à la globine (constituant protéique). L'organisation de la globine en dimères semblables (tétramérie) dont la composition en chaînes polypeptidiques est variée, permet de classer les différents types d'hémoglobines. Les chaînes élémentaires de la globine sont nommées alpha, bêta, delta ou gamma à l'instar des gènes qui codent pour leurs synthèses respectives. L'hémoglobine adulte est un mélange de trois types moléculaires:
| Types d'hémoglobines | Chaînes de globine | Proportion (%) |
| Hémoglobine A normale (HbA) | α2,β2 | 97 - 99 |
| Hémoglobine A2 (HbA2) | α2,α2 | 3,5 |
| Hémoglobine fœtale (HbF) | α2,γ2 | traces |
Tab. 1. Les différents types d'hémoglobines présentes chez l'adulte sain [9].
L'HbA est prépondérante. L’HbF n'existe normalement qu'à l'état de traces chez l'adulte.
2. - Les gènes de l'hémoglobine:
Ils sont situés au niveau de deux chromosomes:
- le chromosome 11 qui porte les gènes β, δ et γ [16].
- le chromosome 16 qui porte le gène α, lequel est dupliqué à l'image du gène γ sur le chromosome 11.
II. - L'HÉMOGLOBINE DRÉPANOCYTAIRE
1. - La mutation drépanocytaire
La drépanocytose résulte de la mutation du gène β-A normal en un allèle récessif anormal β-S. Le gène muté β-S est à l'origine de la production d'une hémoglobine anormale S, qui intervient par sa quantité et ses propriétés dans les manifestations cliniques et biologiques de la drépanocytose.
La maladie se transmet sur le mode autosomique codominant, ce qui explique la double présence des hémoglobines A et S chez les drépanocytaires (le gène normal ne masque pas l'expression moléculaire du gène muté récessif).
On distingue deux formes cliniques:
- la forme hétérozygote ou trait drépanocytaire. Peu symptomatiques, les porteurs du trait drépanocytaire conditionnent la pérennité de la tare en zone d'endémie.
- Les formes homozygotes sont en revanche affectées par une lourde mortalité dans l’enfance.
Il existe des associations hétérozygotes de la drépanocytose avec une thalassémie bêta (β-Thal) ou l'hémoglobinose C (HbC). Du point de vue de la symptomatologie clinique, ces doubles hétérozygoties sont comparables à la drépanocytose homozygote et se regroupent avec elle sous le concept de maladie drépanocytaire.
2. - Propriétés physico-chimiques de l'HbS :
La substitution d'un acide glutamique par une valine dans la structure de l'HbS crée des modifications conformationnelles sur la surface de la molécule (études par résonance magnétique nucléaire). Les modifications siègent aux extrémités N et C-terminales de la chaîne β, expliquent les caractéristiques physiques de l'HbS (moindre solubilité, instabilité mécanique, polymérisation en milieu désoxygéné) et une migration électrophorétique particulière.
2.1. - Hyposolubilité :
Elle s'observe en milieu fortement salin; la désoxy-HbS devient alors 50 fois moins soluble que l'hémoglobine normale HbA.
2.2. - Instabilité mécanique :
L'HbS oxygénée soumise à. une agitation prolongée précipite plus rapidement que l'oxy-HbA [5]. Loin d'être spécifique de l'HbS, cette propriété n'est en pratique pas exploitée.
2.3. - Polymérisation de la désoxy-HbS :
La polymérisation de l'hémoglobine S désoxygénée est à l'origine de la déformation du globule rouge drépanocytaire. Elle se produit aussi bien in vitro en solution concentrée que in vivo au sein de la circulation.
Sous l'effet de la désoxygénation, les molécules d'hémoglobine S s'agrègent en longs polymères ou tactoïdes (fig. 1). In vivo, cette modification structurale de la désoxy-HbS provoque à l'intérieur du globule rouge le passage de l'état fluide à l'état para-cristallin.
Les polymères de désoxy-HbS correspondent à des structures cristallines allongées, en aiguilles, qui emprisonnent des molécules d'hémoglobine encore solubles; l'ensemble prend alors la consistance d'un gel. La cellule est déformée et distendue par ces cristaux qui la traversent de part en part, induisant des lésions membranaires qui diminuent la plasticité érythrocytaire et modifient sa perméabilité. Il en résulte une perturbation des échanges ioniques transmembranaires [48].
La polymérisation de l'HbS est réversible, puis devient irréversible après plusieurs cycles de désoxygénation-réoxygénation. En conséquence, certaines cellules restent définitivement déformées: ce sont alors des drépanocytes irréversibles.
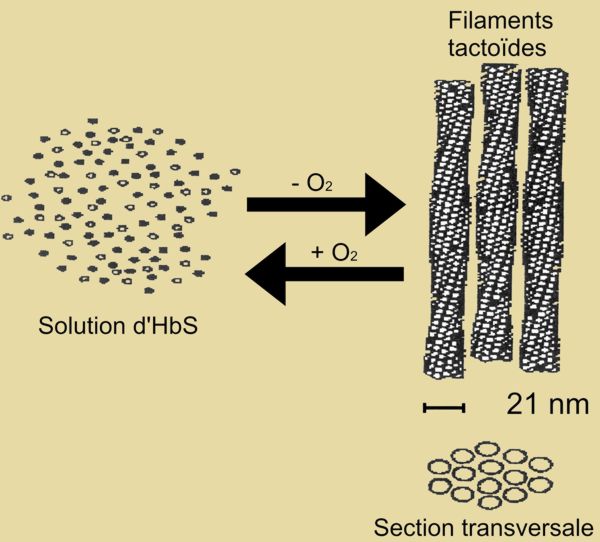
Fig. 1. Formation réversible de filaments tactoïdes sous l'effet de la désoxygénation. [64]
La gélification est en revanche inhibée par l'oxygénation et par la présence de certaines hémoglobines non-S (A, F, C). La présence conjointe d'autres variantes anormales d'hémoglobine baisse le seuil de falciformation; ce sont les hémoglobines D-Los Angeles (β121 : Glu → Gln), O-Arab (β121 : GIu → Lys), Korle-Bu (β373 : Asp → Asn), C-Harlem (β6 : GIu → Val, β73 : Asp → Asn) et C-Memphis (β6 : GIu → Val, α23 : GIu → Gln) [64].
La gélification de l'hémoglobine S se produit après un intervalle de latence [39] qui varie selon les conditions expérimentales; elle est tributaire de la concentration en désoxy-HbS, est favorisée par de nombreux facteurs dont les plus fréquents sont l'élévation de la température, la baisse du pH (acidose) et du taux en acide 2,3-diphosphoglycérique (2,3-DPG), l'hypoxie ou la déshydratation cellulaire.
La déformation cellulaire est polymorphe; elle dépend du mode de cristallisation des fibres et de la résistance membranaire. La présentation classique en faucille est retrouvée si le développement de longs tactoïdes rencontre une structure membranaire souple. La cellule se trouve moins déformée quand prédominent des filaments courts [52]; les cellules denses à membrane rigide apparaissent plus petites et ovalaires. Chez un sujet homozygote, toutes les cellules présentent un certain degré de polymérisation.
3. - Physiopathologie de la drépanocytose
3.1. - Hyperviscosité sanguine :
La drépanocytose apparaît véritablement comme une maladie rhéologique au cours de laquelle il existe une tendance à l'hyperviscosité et à la stase sanguine. La cellule drépanocytaire y contribue par sa forme et des modifications membranaires qui accroissent son adhésivité à la paroi des vaisseaux.
D'autres facteurs extrinsèques au globule rouge agissent aussi en faveur d'une augmentation de la viscosité sanguine; ce sont des facteurs cellulaires et plasmatiques. Entre deux crises de falciformation, l'anémie et le taux bas d'hématocrite tendent à diminuer la viscosité sanguine. Celle-ci augmente en revanche au cours de la crise vaso-occlusive, sous l'influence de certains nombre de facteurs:
- Les drépanocytes: ils interviennent par leur forme et leurs particularités mécaniques. Les drépanocytes majorent d'autant plus la viscosité qu'ils sont au stade d'irréversibilité morphologique. Du point de vue expérimental, ils adhèrent plus facilement â l'endothélium vasculaire que les globules rouges normaux.
- Les réticulocytes: du fait de l'anémie drépanocytaire qui est du type périphérique, la médullopoïése est en permanence active dans la production de réticulocytes, selon un mécanisme de compensation. Les réticulocytes sont des cellules jeunes et de taille plus importante que l'érythrocyte mâture. Par leur nombre et leur taille, ils contribuent à augmenter la viscosité du sang au cours d'une crise avec régénération médullaire accrue.
- Le fibrinogène: en réponse au stress vaso-occlusif [44], le taux de fibrinogène augmente au cours d'une crise et avec lui la viscosité; l'élévation du taux de fibrinogène s'observe même en l'absence d'une participation infectieuse [58].
- Les plaquettes: il y a une activation plaquettaire en période de crise comme en témoignent sur le plan expérimental l'augmentation d'adénosine triphosphate (ATP) circulant et la baisse du taux d'adénosine diphosphate (ADP) intra-plaquettaire [11]; l'activation des plaquettes a pour conséquence l'augmentation de leur agrégabilité.
- Les cellules endothéliales circulantes: en période critique l'augmentation du nombre de cellules endothéliales circulantes rend compte d'importantes lésions vasculaires par anoxie [66].
3.2. - Hypercoagulabilité sanguine :
- La protéine C: c'est une glycoprotéine plasmatique, d'origine hépatique et vitamine K dépendante. La forme activée de cette hétéroprotéine (Ca) agit comme un anticoagulant fibrino-freinateur. L'activation de la protéine C en Ca est le fait du complexe thrombine-thrombomoduline.
La Ca nécessite l'intervention d'un cofacteur, la protéine S, pour exercer une lyse enzymatique sur les formes activées de l'accélérine (Va) et du facteur antihémophilique (VIIla), ce qui interrompt la chaîne de la fibrinoformation.
Le déficit partiel en protéine C expose au risque de thromboses veineuses à répétition. Si le déficit est complet, les accidents thrombotiques sont précoces (50% avant l'âge de 30 ans) [43]. Le taux de protéine C est abaissé pendant la crise vaso-occlusive drépanocytaire et peut être à l'origine d'accidents thrombotiques. Cette baisse provient d'un dysfonctionnement hépatique pendant la crise (troubles de la circulation intra-hépatique, thromboses au niveau des sinusoïdes hépatiques et engorgement des cellules de Kupffer par des drépanocytes phagocytés).
- L'antithrombine III (AT-III): les malades drépanocytaires présentent un déficit en AT-III [43]. L'antithrombine III est un inhibiteur physiologique de la stabilisation de la fibrine en caillot définitif. Elle est en fait le substrat préférentiel de la thrombine et ce n'est que dans le cas où l'AT-III disponible est saturée que la thrombine peut s'attaquer au fibrinogène. L'antithrombine III et la protéine C agissent donc en complémentarité pour limiter la fibrinogénèse. Leur baisse conjointe au cours de la crise drépanocytaire majore le risque d'accidents thromboemboliques.
III. - CONSÉQUENCES CLINIQUES :
Hyperviscosité sanguine et hypercytose font de la drépanocytose une maladie rhéologique où prédominent des phénomènes thromboemboliques. Le risque d'accidents thromboemboliques est favorisé au cours d'une crise, par la baisse de deux protéines anticoagulantes importantes que sont la protéine C et l' antithrombine III.
Les manifestations douloureuses des périodes critiques témoignent de phénomènes ischémiques au niveau tissulaire, dus à des microthromboses multiples.
![]()
![]()
![]()
Chapitre III : ÉPIDÉMIOLOGIE
I. - RÉPARTITION GÉOGRAPHIQUE
150 000 enfants drépanocytaires naissent par an dans le monde, dont 120 000 en Afrique [28]; la plupart sont homozygotes pour l'hémoglobine S, mais on rencontre aussi des associations hétérozygotes avec une thalassémie bêta ou l'hémoglobinose C. La démographie drépanocytaire est de répartition inégale:
- En Afrique: la drépanocytose connaît une prévalence maximale en Afrique sub-saharienne et se retrouve sur tout le continent. La zone de prévalence drépanocytaire africaine correspond à la bande géographique comprise entre le 15e parallèle de latitude nord et le 20e parallèle de latitude sud: c'est la ceinture sicklémique (sickle belt) définie par LEHMANN et HUNTSMAN en 1974. Dans certaines populations africaines et selon des données relativement anciennes, 20 à 40% des sujets sont porteurs du trait drépanocytaire [21]. L`hémoglobinose C se rencontre principalement au Burkina-Faso (ex Haute-Volta), dans le nord ghanéen et à un degré moindre dans le nord-est du Nigeria. La bêta-thalassémie se rencontre en Afrique associée à la drépanocytose au Liberia (S/β+-Thal) et dans le Nord saharien (S/β0-Thal) [28].
- Aux Etats-Unis d'Amérique: la mutation drépanocytaire est la plus répandue des anomalies de l'hémoglobine. Dans le sud de ce pays, environ 7% de la population noire sont porteurs du trait drépanocytaire [62]. Le gène est également retrouvé de façon sporadique dans les autres groupes ethniques. L'hémoglobinose C est retrouvée chez les Noirs américains dans les proportions de 2 à 3%. Les associations hétérozygotes cliniquement symptomatiques (S/C et S/β+) sont également présentes.
D'une manière générale, le gène drépanocytaire affecte tous les pays de grande migration noire africaine: Etats-Unis d'Amérique, Antilles, Brésil.
- Les pays du bassin méditerranéen: la maladie drépanocytaire se rencontre à un moindre degré dans tout le bassin méditerranéen: Afrique du nord, la péninsule ibérique, la Sicile, l'Italie du sud, la Grèce, la Turquie et le Proche-Orient. On y retrouve surtout la mutation bêta-thalassémique1.
II. - DRÉPANOCYTOSE ET PALUDISME
L'interaction drépanocytose-paludisme s'explique par une certaine protection concédée par l'HbS contre le parasite paludéen, s'agissant du Plasmodium falciparum pour lequel cet aspect est mieux connu et largement documenté. La protection concerne les seuls porteurs du trait drépanocytaire et leur procure un avantage sélectif comparativement aux sujets normaux [51].
Le comportement du parasite paludéen au sein de l'hématie S se prête à l'observation expérimentale et tout se passe comme si l'hémoglobine drépanocytaire était un substrat peu propice à sa maturation: en effet, confronté à l'HbS, le Plasmodium devient peu viable. Des observations en microscopie électronique mettent en évidence des signes de souffrance parasitaire (ruptures membranaires provoquées par les filaments d'hémoglobine gélifiée, vacuolisations internes) [30, 51].
D'autres observations ont révélé des modifications membranaires au niveau du globule rouge S parasité, qui sont responsables d'une accélération de sa falciformation et d'une adhésivité accrue à la paroi endothéliale lorsque la tension en oxygène baisse.
Le Plasmodium paludéen voit son développement compromis en présence d'hémoglobine S et est rapidement éliminé du sang des hétérozygotes, qui bénéficient alors d'une moindre mortalité due au parasite.
Au stade de la circulation profonde, l'hématie drépanocytaire parasitée rencontre les conditions idéales pour falciformer (stase circulatoire, acidose relative et bas taux d'O2) et être éliminée avec son bagage parasitaire, au prix incontournable d'une anémie mieux supportée par les hétérozygotes. La relation drépanocytose-paludisme appelle un autre type d'observation: loin des zones endémiques d'Afrique, les enclaves européennes de la malaria (Grèce, Italie) sont aussi celles où le gène β-S est retrouvé [3].
La démographie drépanocytaire paraît donc tributaire de l'existence passée ou présente de l'endémie palustre sur un site géographique donné. La ceinture sicklémique elle-même coïncide avec une zone africaine de forte endémie palustre [3].
L'endémie palustre est donc un facteur important de maintien de la drépanocytose, puisqu'elle sélectionne des hétérozygotes asymptomatiques qui transmettront durablement le gène β-S.
La relation drépanocytose est bidirectionnelle: le paludisme peut exister chez certains sujets, en portage asymptomatique pour le parasite. Ce portage muet est un facteur reconnu dans la majoration de la symptomatologie drépanocytaire [33]. A partir de cette observation, la protection apportée par le paludisme respecte donc une certaine nuance en conservant toutefois son intérêt épidémiologique.
III. - PROPAGATION MONDIALE DU GENE DRÉPANOCYTAIRE
La drépanocytose est prépondérante en Afrique dans ses diverses configurations génétiques (homozygotes et doubles hétérozygotes malades d'une part, porteurs sains d'autre part). Cela signifie au moins que l'origine de la mutation drépanocytaire est africaine. Cependant, la maladie n'apparaît plus aujourd'hui comme une pathologie spécifique à l'Afrique et aux populations noires car l'accroissement des migrations internationales, les modifications du comportement social avec les mariages inter-raciaux élargissent la portée démographique et géographique de la maladie.
Aussi, les données épidémiologiques actuelles présentent-elles la drépanocytose comme une maladie mondialement rencontrée, transposée depuis l'Afrique aux autres pays du monde selon une distribution inégale.
La drépanocytose manifeste sa présence au niveau mondial [55]. La transmission inter-raciale d'une maladie génétique demeurant toujours possible, Il est devenu plus judicieux de préférer provisoirement la notion de d'électivité ethnique à celle de la spécificité raciale. Car selon toute vraisemblance, une approche épidémiologique fondée uniquement sur le seul critère racial pourrait s'avérer insuffisante d'ici quelques générations.
L'implantation du gène β-Sen Amérique du nord et au Portugal est principalement le fait de migrations africaines dans des contextes historiques connus. Le fait de retrouver des sujets porteurs du gène (ou malades) dans d'autres groupes ethniques trouve une explication dans les croisements inter-ethniques et fournit donc des indications sur l'histoire génétique des personnes. La succession des générations et la dilution des particularités culturelles encouragent à prendre comptes d'autres arguments que le seul phénotype physique.
Le développement actuel des voyages internationaux favorise le déplacement des populations et vient inclure la drépanocytose au répertoire des maladies mondialement rencontrées; les populations, immigrées se fixant par la suite durablement en assurant une descendance, le gène drépanocytaire tend à devenir ubiquitaire, forçant d'une certaine manière les pays hôtes à se familiariser avec des préoccupations sanitaires nouvelles amenées par une pathologie initialement importée.
Et si l'on prend en considération l'association possible de la drépanocytose à d'autres hémoglobinopathies héréditaires (thalassémie bêta et hémoglobinose C), il est prévisible que la maladie drépanocytaire posera des problèmes de plus en plus importants en santé publique.
En France métropolitaine, la répartition des hémoglobinopathies suit celle des populations immigrées originaires des pays à prévalence élevée d'hémoglobinopathies héréditaires. D'après le recensement de 1982 vivaient en France 3,5 millions d'immigrés, dont 2,66 millions (76 %) étaient originaires de ces pays «à risque».
Ces chiffres sont vraisemblablement sous-estimés car les dépistages ne sont ni systématiques ni généralisés.
En mai 1991, 1 279 syndromes drépanocytaires majeurs (SDM) ont été répertoriés en France métropolitaine dont 855 en région parisienne (67 %) et 424 en province (33 %) (Fig. 2). [47].
En Île-de-France, la drépanocytose constitue le premier risque génétique avec un enfant homozygote sur mille (projection réalisée à partir de quelques grandes maternités du Val-de-Marne et de Paris). Chez les nouveau-nés de la région parisienne, les hémoglobinopathies sont devenues au moins trois fois plus fréquentes que la mucoviscidose et dans certains services de pédiatrie, environ 10 % des lits sont occupés par des enfants drépanocytaires [32].
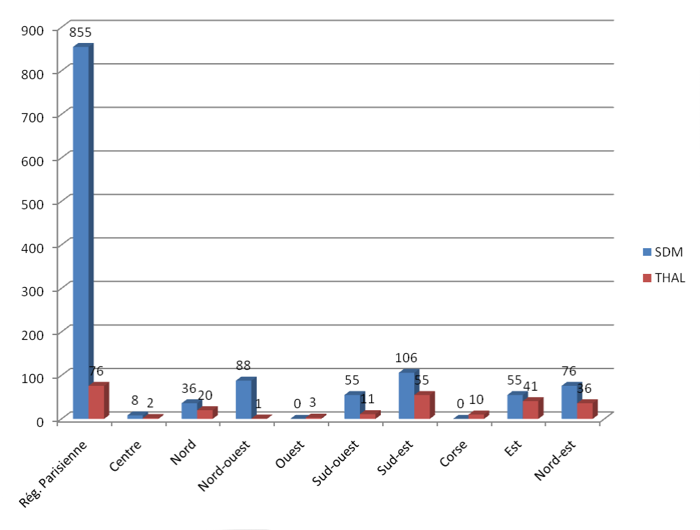
Fig. 2. Répartition des β-thalassémies majeures (THAL) et des syndromes drépanocytaires majeurs (SDM) en France métropolitaine en 1991 [47].
| β-thalassémie homozygote | Population à risque | |
| Région parisienne | 29,8 % | 36,5 % |
| Province | 70,2 % | 63,5 % |
| Cas répertoriés | 1279 | |
| Nombre (31.10.91) | 2'808'330 (recensement 1982) |
Tab. 2. Répartition géographique des malades atteints de b-thalassémie homozygote et des populations originaires des pays à haut risque pour cette pathologie [47].
La fréquence du gène β-S est devenue importante dans toutes les grandes villes européennes (Londres, Paris, Bruxelles). Les unions matrimoniales interraciales sont devenues courantes et cela peut à long terme modifier les données épidémiologiques actuellement connues, avec des sujets drépanocytaires faisant partie d'une génération qui ne répondra plus aux caractéristiques retenues jusqu'aujourd'hui.
| SDM | Population à risque | |
| Région parisienne | 67 % | 61 % |
| Province | 33 % | 39 % |
| Cas répertoriés | 241 | |
| Nombre (31.10.91) | 647'713 (Antilles, Afrique noire) |
Tab. 3. Répartition géographique des malades atteints de syndrome drépanocytaire majeur et des populations originaires des pays à haut risque pour cette pathologie [47].
Des cas sporadiques de drépanocytose-maladie sont relatés aux Etats-Unis d'Amérique dans des groupes sociaux non noirs, c'est-à-dire blancs, orientaux ou indiens d'Amérique, dépistés à l'occasion d'un tableau douloureux avec anémie et pâleur, ou à titre systématique. Leurs génotypes sont aussi variés que ceux rencontrés en Afrique (S/S, S/β+ ou S/C) [60]. Concernant les sujets de ces observations tirées de la littérature, rien ne laissait supposer sur le plan phénotypique leur ascendance noire.
En conclusion il convient devant une anémie hémolytique congénitale non diagnostiquée, de prendre en compte la drépanocytose comme diagnostic d'élimination, sans donner de valeur absolue, dans certains cas aux critères ethniques, raciaux ou géographiques.
Notre expérience personnelle en pratique de ville nous conforte dans cette idée: l'interrogatoire des parents d'un enfant métis adopté nous a permis de vérifier l'existence d'une anémie modérée, la notion d'un traitement martial antérieur pour anémie, des antécédents de douleurs abdominales à répétition, qui coïncidaient avec des situations particulières (bronchites, altitude).
L'électrophorèse de l'hémoglobine a apporté la conclusion d'une drépanocytose hétérozygote avec les résultats suivants2:
HbA1: 52,10 % (N > 96,5)
HbA2: 2,90 % (1,50 < N < 3,50)
HbS: 40,7 %.
HbF : 4,3 % (N < 1)
Le pronostic évolutif est ici optimiste. Il s'agit néanmoins d'un cas d'hétérozygotisme symptomatique dépisté. Par la suite, ce portage hétérozygote deviendra particulièrement important à considérer à l'âge nuptial, en fonction des origines du futur conjoint.
Les pays médicalement développés et sensibilisés aux problèmes de la maladie drépanocytaire sont actuellement responsables d'une amélioration notable tant de l'espérance que du confort de vie des malades. Paradoxalement, en dehors des services hospitaliers spécialisés, la drépanocytose reste assez mal connue dans la pratique médicale courante.
IV. - POLYMORPHISME GÉNÉTIQUE DE LA DRÉPANOCYTOSE
Si les propriétés physico-chimiques de l'hémoglobine S sont identiques et ne dépendent pas de l'origine des malades considérés, en revanche, à l'intérieur d'un même pays, des différences dans l'expressivité clinique de la maladie peuvent se retrouver si on compare des groupes ethniques différents [23].
S'adressant à des populations apparemment homogènes et au sein d'une même famille, il arrive que l'on observe des degrés de sévérité variables pour la drépanocytose. C'est également le cas dans les comparaisons établies entre différents pays. L'expressivité et la sévérité clinique de la maladie varient d'un groupe à un autre et, à l'intérieur d'un même groupe, d'un individu à l'autre.
Des complications spécifiques apparaissent dans certains pays qui sont absentes dans d'autres [2, 15]. À titre d'exemple, il existe des différences tant hématologiques que cliniques entre les drépanocytaires du Sénégal d'une part et ceux du Bénin et de la République Centrafricaine, d'autre part [64].
Au Sénégal, on observe moins de cellules denses et de drépanocytes irréversibles (donc moins de falciformation); le taux d'hémoglobine fœtale est relativement élevé, ce qui laisse présager d'un pronostic évolutif plus optimiste. La drépanocytose est de sévérité clinique décroissante quand on considère respectivement la République Centrafricaine, le Bénin et le Sénégal.
Le postulat résultant de ces constatations a été de chercher à établir une relation entre les différences observées sur le plan clinique et des configurations génétiques qui seraient variables d'un groupe à un autre.
Les études se sont orientées vers l'ADN de la région génomique 13 portée sur le bras court du chromosome 11, sur un échantillon humain composé de 46 noirs (A/A, A/S, S/S), 27 caucasiens et asiatiques tous normaux (A/A) [42].
Explorant cette région génomique à l'aide d'une enzyme de restriction, l'Hpa 1, KAN et DOZY (1978) ont décrit un polymorphisme situé à 5 kilo bases (Kb) du gène β.
L’endonucléase Hpa 1 reconnaît la séquence G-T-T-A-A-C et y opère un clivage; une mutation de cette séquence empêche la restriction enzymatique. La succession séquence non mutée (clivage) et séquence mutée (absence de clivage) correspond à un contexte génomique particulier ou haplotype. La variabilité des haplotypes définit un polymorphisme (Fig. 4).
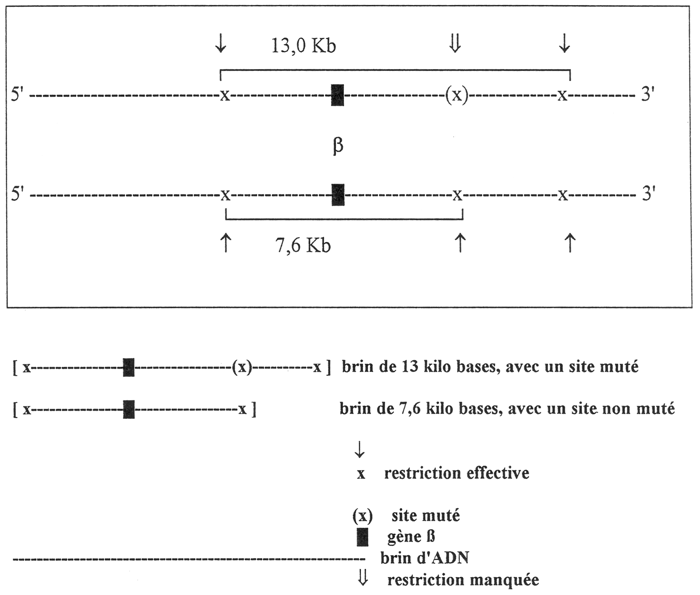
Fig. 4. Restriction enzymatique Hpa 1 sur d'ADN du chromosome 11: les brins de longueurs identiques ont des haplotypes comparables.
Lorsque le site de restriction enzymatique manque, les fragments d'ADN sont plus longs (13 Kb) que si le site est présent (7,6 Kb). D'autres variantes ont été retrouvés avec des fragments de 7 Kb. Les fragments de 7,6 et 13,0 Kb sont tous les deux retrouvés chez les sujets de l'étude qui étaient d'origine africaine. Le fragment de 13,0 Kb s'est retrouvé fréquemment associé à la mutation drépanocytaire pour cette étude américaine, de même que lors d'une étude similaire menée en Afrique. Il faut ensuite transposer ces résultats au niveau de l'expression phénotypique de la drépanocytose pour rechercher sinon expliquer les différences physiopathologiques et donc cliniques qui seraient commandées par des présentations génétiques différentes. Autrement dit, deux gènes β-S ayant un environnement chromosomique identique peuvent-ils générer des situations cliniques comparables?
La réponse à cette question est rendue difficile au moins par la multiplicité d'autres facteurs dont les rôles sont plus évidents à établir dans la modulation de la symptomatologie drépanocytaire: la différence fondamentale entre le drépanocytaire américain et son homologue africain ne relève certainement que de la qualité du suivi médical, variable d'un pays à l'autre.
Chez les Noirs américains, le site Hpa 1 manque dans 60 à 80 % chez les porteurs du gène β-S, alors qu'il est majoritaire chez les porteurs du gène β-A normal.
Des études similaires menées en Afrique sur des points géographiques différents et intéressant des groupes ethniques variés ont démontré que la relation entre le gène β-S et l'absence su site Hpa 1 (brins de 13,0 Kb) sont propres aux groupes ethniques du golfe du Bénin et, sans doute par le fait de migrations transsahariennes, aux groupes d'Afrique du nord [50].
La mutation du site de restriction enzymatique n'accompagne pas nécessairement le gène β-S. Il y a par ailleurs une dissémination des résultats obtenus. Au Sénégal et en pays bantou sont retrouvés des brins de 7,6 Kb. Enfin, en pratique, trois haplotypes se distinguent par leur prévalence: Sénégal, Bantou (ou CAR3), Bénin [64].
| Type de brin (kb) | Gène associé | |
| Algérie | 13,0 | β-S |
| Maroc | 13,0 | β-S |
| Côte d'Ivoire | 7,6 | β-S (72 %) |
| 13,0 | β-S (28 %) | |
| 13,0 | β-S (18 %) | |
| 13,0 | β-A (100 %) | |
| Togo | 13,0 | β-A (17 %) |
| 7,6 | β-S | |
| Gabon | 7,6 | β-S |
| Kenya | 7,6 | β-S |
| Arabie Saoudite | 7,6 | β-S |
Tab. 4. Distribution géographique du polymorphisme Hpa 1. [50]
L'étude du polymorphisme génétique de la drépanocytose renseigne principalement sur l'origine de la mutation drépanocytaire: un haplotype donné, retrouvé en Amérique, a-t-il son correspondant en Afrique? La réponse est en pratique affirmative dans certains cas, mais pas toujours lorsque la question est de savoir si la présentation clinique est identique pour deux haplotypes identiques présents autour de la mutation drépanocytaire et considérés à des lieux géographiques différents.
La drépanocytose paraît bénigne au Sénégal, en Inde, en Arabie Saoudite et au Zaïre alors qu'ailleurs, le décès précoce des malades est une constante. C'est le polymorphisme d'expression, dont les facteurs de modulation sont mieux connus:
- facteurs socio-économiques permettant ou non une couverture médicale suffisante.
- facteurs culturels et climatiques avec la question des multiples pathologies associées qui modifient le cours de la drépanocytose [65].
- facteurs génétiques.
V. - FACTEURS GÉNÉTIQUES DE MODULATION DE LA DRÉPANOCYTOSE
1. - Les alpha-thalassémies :
La production des chaînes alpha de la globine incombe au gène α, localisé sur le chromosome 16; ce gène est généralement dupliqué dans l'espèce humaine.
Les α-thalassémies résultent habituellement d'une délétion en nombre variable de ces gènes; aussi, les formules génotypiques rencontrées s'étendent-elles des formes hétérozygotes mineures [-α/α, α], [-α/-α] et [--/αα] à la forme homozygote létale [--I--], fréquente en Asie du sud-est.
L'hétérozygotisme [-α/αα] est fréquente dans les populations noires africaines et s'exprime le plus souvent par une microcytose isolée [38]. Chez le thalassémique alpha porteur d'une mutation β, le déficit en chaîne a provoque la baisse relative du constituant anormal S ou C par association préférentielle des chaînes déficitaires avec les chaînes β non mutées [13, 40]. Le taux d'HbS chez l'homozygote S/S décroît avec l'importance d'une α-thalassémie associée [73]. L'α-thalassémie semble de cette façon procurer aux drépanocytaires homozygotes une survie prolongée.
2. - L'hémoglobine fœtale (HbF) :
Le rôle modérateur de l'hémoglobine fœtale dans la symptomatologie drépanocytaire est connu depuis 1961. Certains sujets homozygotes pour l'hémoglobine S à l'électrophorèse, n'étaient pas pour autant anémiques et ne souffraient pas de crises drépanocytaires typiques [41]. Ces sujets présentaient à l'électrophorèse un taux exceptionnellement élevé d'hémoglobine fœtale.
La polymérisation de l'HbS dépend de sa concentration intraérythrocytaire. L'HbF ne copolymérise pas avec l'HbS et donc, la substitution dans une cellule d'une quantité d'HbS par de l'HbF baisse la concentration en hémoglobine polymérisable.
Chez le nouveau-né S/S, la polymérisation n'apparaît que lorsque l'HbF diminue; chez les rares hétérozygotes S/PHHF4 la polymérisation est inexistante. Et dans certaines populations d'Arabie Saoudite on retrouve chez des malades S/S une production pancellulaire d'HbF à des taux élevés [26, 55], d'où une moindre expressivité de la drépanocytose même dans sa présentation homozygote.
3. - L'hémoglobine A2 (HbA2) :
D'après Van Ros5, le taux d'HbA2 est généralement élevé (1,9 à 5 %) au cours de la drépanocytose homozygote, mais dans des proportions moins importantes que chez les thalasso-drépanocytaires. Il semble exister une corrélation entre la gravité de la drépanocytose et le taux d'HbA2: des taux élevés d'HbA2 atténuent la symptomatologie drépanocytaire même en l'absence d'une majoration conjointe de l'HbF [34].
L'HbA2 intervient dans la sévérité de la drépanocytaire en diluant l’HbS, autrement dit en réduisant de manière relative la fraction d'hémoglobine polymérisable S.
4. - Les hémoglobines anormales autres que l'HbS :
Les associations hybrides d'autres hémoglobines anormales avec l'HbS sont fréquentes en Afrique. Lorsqu'elles copolymérisent avec l'hémoglobine S, ces hémoglobines stabilisent ou déstabilisent selon leur nature, la liaison entre les fibres d'hémoglobine gélifiée.
Dans certains cas, l'interaction déstabilisant le polymère est bénéfique (S/Korle-Bu, S/D-Lepore).
(+)
| HbS/HbS |
| HbS/Hb C Harlem |
| HbS/Hb C |
| HbS/Hb O Punjab |
| HbS/Hb O Arab |
| HbS/HbA |
| HbS/Hb Korle Bu |
| HbS/Hb Lepore-Boston |
(-)
Fig. 4. Echelle des risques de falciformation en fonction de l'hémoglobine interagissant avec l'HbS.
A l'opposé, d'autres associations stabilisent le polymère (S/D-Punjab, S/OArab). Il en découle que selon le type d'hybride moléculaire, la polymérisation de l'hémoglobine ainsi que sa réversibilité sont plus ou moins faciles [71].
5. - Le chromosome X :
Plus récemment, le rôle du chromosome féminin a été évoqué sur la base d'une expression de l'hémoglobine fœtale variable selon le sexe. Cette différence joue au niveau de la production, génétiquement déterminée, des cellules F6.
Ce déterminisme génétique serait supporté par le chromosome X, et permettrait la production d'une population de cellules souches F plus actives chez les filles [45]: il en résulterait alors une production d'hémoglobine fœtale plus importante chez ces dernières. L'HbF n'étant pas systématiquement augmentée chez toutes les filles, on pense que ce prédéterminisme génétique est modulé par des facteurs ethniques.
![]()
![]()
![]()
Notes
- La β-thalassémie est une anomalie quantitative constitutionnelle de l'hémoglobine. De transmission autosomique récessive, elle conduit soit au défaut complet de la synthèse des chaînes β de la globine, soit à une simple réduction de cette synthèse.
- Les normes et notations sont celles du laboratoire d'analyses médicales Notre-Dame, Amiens.
- CAR: Central African Republic.
- PHHF: Persistance Héréditaire de l'Hémoglobine fœtale.
- VAN ROS G.: genetic and clinical forms of sickle-cell syndromes in Zaïrians Soc belge Med Trop 1975, 55, 609-622.
- Les cellules F contribuent à l'hématopoïèse par la synthèse des chaînes γ qui entrent dans la constitution de l'hémoglobine fœtale.
SOMMAIRE | HISTORIQUE | RAPPELS | ÉPIDÉMIOLOGIE |ÉTUDE CLINIQUE | DIAGNOSTIC POSITIF DE LA DRÉPANOCYTOSE | DIAGNOSTIC ANTE ET NÉONATAL | TRAITEMENT DE LA DRÉPANOCYTOSE | LES PERSPECTIVES THÉRAPEUTIQUES DE LA DRÉPANOCYTOSE | CONCLUSION | BIBLIOGRAPHIE